Department of Anatomy and Structural Biology, University of Otago, Dunedin, New Zealand
The subarachnoid space consists of a number of distinct compartments called subarachnoid cisterns. Knowledge of cisternal anatomy is very important not only for anatomists but also for clinicians, particularly neurosurgeons. This paper reports a technique which combines the traditional E12 sheet plastination method with several special treatments so that the subarachnoid space, transcisternal arteries and veins, cranial nerves and arachnoid trabeculae are preserved in a relatively natural state and shown with different colours. This technique should greatly facilitate cisternal anatomy studies and provide a new approach for examining structures in the subarachnoid space at both macroscopic and microscopic levels.
subarachnoid space, sheet plastination, cisternal anatomy
Dr Ming Zhang, Department of Anatomy and Structural Biology, University of Otago, PO. Box 913, Dunedin, New Zealand. Telephone: 64 3 479 7378 / Fax: 64 3 479 7254 Email: zhang.ming@stonebow.otago.ac.nz

![]()



Compartmentalization of the subarachnoid space (SAS) into subarachnoid cisterns by arachnoid trabecular walls has been widely described (Yasargil et al., 1976; Matsuno et al., 1988; Brasil and Schneider, 1993; Vinas et al., 1994,1996a,b). Most of the intracranial operations for intracranial aneurysms, brain tumors and disorders of cranial nerves are directed through the subarachnoid cisterns. These cisterns provide a natural pathway through which the major intracranial arteries, veins and cranial nerves can be approached. Thus cisternal anatomy (the anatomical relationship of arachnoid trabecular walls with vascular and neural elements) has significant importance not only to anatomists but also to clinicians, especially neurosurgeons.
Current knowledge of cisternal anatomy mainly comes from two sources: anatomical dissection and clinical observation, such as radiological examination and intracranial operation. However, the fine and delicate arachnoid trabeculae are easily destroyed in anatomical dissection when brain is removed from its cranial cavity during preparation. Also, pathological changes in the brain may affect clinical observation. Thus, in order to obtain reliable information about cisternal anatomy, a method that can preserve the SAS and its contents in a natural state without above drawbacks is needed.
Sheet plastination is a recently developed technique in which water and lipids of tissues are replaced by curable resin on a cellular level. The sheet plastination technique has been widely applied to human brain studies to demonstrate neuroanatomy (see Grondin and Olry, 1996 for review). However the subarachnoid cisterns and their contents can not be adequately demonstrated by this technique. In this study, we have modified the traditional techniques so that the SAS, arachnoid membranes, transcistemal arteries and veins can be preserved in a natural state and stained with different colors. Our modified technique should greatly facilitate further cisternal anatomy studies, and provide a new approach for examining structures in the arachnoid cisterns at both macroscopic and microscopic levels.
Materials
Cadavers of three female humans aged from 75 to 85, and 8 sheep were used in this study. The experiments using sheep were approved by the University of Otago Committee on Ethics in the Care & Use of Laboratory Animals.
Fixation
The right femoral artery and vein of a cadaver were
cannulated and blood was washed out by saline. Ten percent formalin followed to fix the whole body. The body was then stored in the same solution until required.
Pretreatment
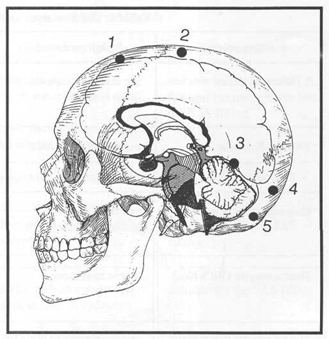
Figure 1. A lateral view of the skull showing the location of the burr holes. On each side, two burr holes on the vault (1&2) cover the frontal and superior regions of the subarachnoid space (SAS) and two burr holes (3&4) above and one (5) below the nuchal line cover the superior and inferior tentorial regions of the SAS, respectively.
Arachnoid mater staining:
In order to completely stain the whole arachnoid mater, ten burr holes of 0.5 mm in diameter were drilled into the skull. The locations of these holes are shown in figure 1. Catheters of 14 gauge were properly inserted into the SAS with the aid of a stylet with dura puncture. The burr holes were then sealed using epoxy glue. Each hole was perfused with 10 ml of Gill's haematoxylin No.2. The SAS was stained for 2-3 minutes and then irrigated using distilled water to wash out the staining solution. The staining was made blue with the Scott's tap water for 2-3 minutes, 40 ml for each hole. Then the SAS was irrigated with the same amount of distilled water.
Vascular fillings:
The internal jugular veins and internal carotid arteries were exposed and cannulated at both sides and perfused with blue and red coloured epoxy resin E20, respectively.
SAS perfusion:
After arachnoid mater staining and vascular filling, the SAS was perfused with 400ml of 10% gelatin solution, 40 ml for each hole. The cadaver was left at room temperature for 24 hours to allow the gelatin to become solid.
Variations for pretreatment
In order to achieve optimal results for staining of arachnoid trabeculae and fillings of vessels and SAS, eight sheep heads were used in our preliminary experiments. Table 1 and Table 2 summarise the materials which were used for the arachnoid mater staining (Table 1) and the SAS and vessel fillings (Table 2).
| Staining solution | Stability of staining | Results | Comments |
| 0.1% cresyl violet | not stable in acetone | the resin becomes milky white due to acidity | the SAS needs to be acidified by 10% acetic acid. |
| 1% toluidine blue | not stable in acetone | the staining fade away during dehydration | |
| Haematoxylin & eosin | eosin staining is not stable in acetone. | blue-stained tissue | |
| Haematoxylin Gill's No.3 | stable in acetone | black blue-stained tissue | hard to control the incubation time as it was less than 1 minute |
| Haematoxylin Gill's No.2 | stable in acetone | purple blue-stained tissue | relatively easy to control |
| Materials | Advantage | Disadvantage | Comments |
| epoxy resin E20 | translucent and similar to E12 | becomes milky white when water and acid exist.
high viscosity and needs to be diluted with acetone |
suitable for the vascular fillings, but not suitable for the SAS filling. |
| 20% gelatine | soluble in water | yellowish after dehydration | not suitable for the SAS filling |
| 20% gelatine with
5% Arabic gum |
soluble in water | becomes a white mass after dehydration | not suitable for the SAS filling. |
| 10% gelatine | transparent and soluble in water | a few web-like structures appear after dehydration | suitable for the SAS filling |
| 5% gelatine | transparent and soluble in water | not solid at room temperature | not suitable for the SAS filling |
E12 sheet plastination
Slicing:
The head of a cadaver was disarticulated at the second cervical vertebra level (C2), embedded in 20% gelatin solution and frozen at -30°C to make a gelatin block. Then the gelatin block was frozen at -80°C for 24 hours. The block was sectioned in a thickness of 2.5 mm by a butcher's bandsaw and the cutting surfaces of slices were cleaned with tap water.
Dehydration:
The slices were laid between mesh and stacked in a grid basket. The basket was immersed in ascending concentration of acetone (95% to 100%) at -30°C. The acetone was replaced every week with higher concentration. The dehydration was completed when the water content in the acetone was less than 1%. The dehydration process took 6 weeks.
Degreasing:
After dehydration, the specimens were left in final acetone bath for degreasing. The temperature of acetone bath was kept at 22-24°C. The degreasing was completed after 2 weeks at which time the fat tissues appeared translucent.
Forced impregnation:
The degreased slices were immersed in a polymer mixture and placed in a vacuum container at 0°C for 36 hours (see "Plastination Workshop", 1997). The polymer mixture was Biodur E12/E1/AE10/AE30 (BIODUR; Rathausstrasse 18, 69126 Heidelberg, Germany) in a ratio of 100:28:20:5 pbw (parts by weight).
Hardening:
The impregnated slices were laid between 0.25mm A.P.E.T (Amorphous Polyethylene Terephthalate, Progressive Plastic Ltd, 31-37 Fraytt St, Dunedin, New Zealand) plastic sheets and cured at 32°C for one week and then placed in an oven at 45°C for another week. Then the oven was switched off and allowed to cool down gradually before the slices were removed.
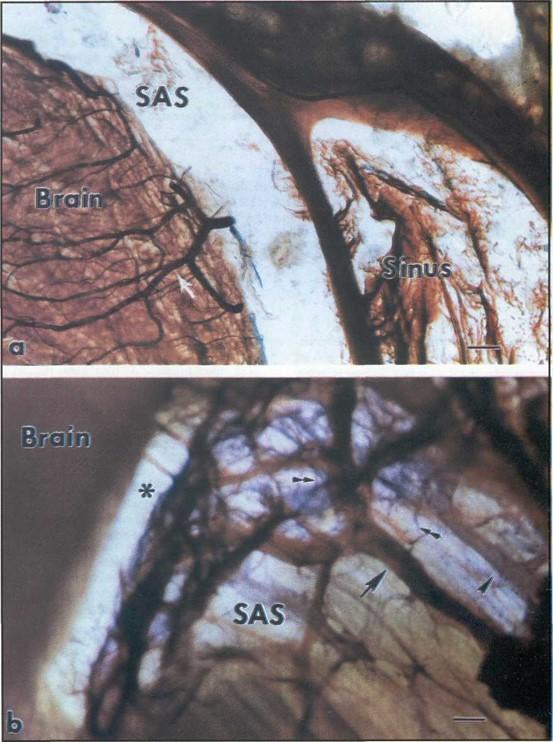
Figure 2. Two plastinated slices prepared with the traditional E12 sheet plastination technique (a) and with the technique developed in this study (b). Bar =1 mm.
(a) shows that the brain tissue shrank, the subarachnoid space (SAS) enlarged, and the vessels in the brain were preserved well (arrow) but the most SAS contents were destroyed.
(b) demonstrates that the transcisternal arteries (arrow), veins (arrowhead), and the arachnoid trabeculae (double arrowheads) were held in their natural positions. The SAS was also well preserved in its natural size although an artifactual gap (asterisk) appeared underneath the pia mater due to the brain tissue shrinkage.
Initial method
The SAS is located between the arachnoid mater and pia mater and mainly contains cranial nerves, arachnoid trabeculae, transcisternal arteries and veins. Our initial method for preserving the SAS and its contents was based on E12 sheet plastination protocol (von Hagens et al.,1987; Cook, 1997). Figure 2a shows a specimen which was processed with a standard E12 sheet plastination technique after the arteries were perfused with red epoxy resin E20. The fine vascular structures were very well preserved in the brain tissue but the SAS and its contents were completely destroyed. It is impossible to distinguish the pia mater, arachnoid mater and arachnoid trabeculae from surrounding structures in the specimen which was processed using the standard sheet plastination technique.
Modified method
In order to overcome the above problems, 8 sheep heads were used in the preliminary experiments. Tables 1 and 2 summarise the results and the recommended procedures have been described in the Methods section. With this modified E12 sheet plastination technique, the SAS (transparent with a few fine yellowish web-like lines), transcisternal arteries (red) and veins (blue), the cranial nerves and the arachnoid trabeculae (purple-blue) were well preserved in a relatively natural state and stained with different colours (figure 2b).
In this study, for the first time, we provide a method for using E12 sheet plastination technique with several special treatments to preserve the SAS, transcisternal arteries and veins, the cranial nerves and the arachnoid trabeculae in a relatively natural state with different colours. Our method should greatly facilitate further cisternal anatomy studies and provide a new approach for examining structures in the SAS at both macroscopic and microscopic levels.
The protocol that we have established is based on a combination of the traditional E12 sheet plastination technique (von Hagens et al. 1987; Cook, 1997) and a technique of SAS perfusion and staining (Brasil and Schneider, 1993). However, there were several limitations for a simple combination. For example, according to the Brasil and Schneider's method, the SAS needs to be acidified in order to stain SAS with cresyl violet. However, the acidification makes translucent epoxy resin become milky white after its injection. Thus the structures in the SAS can not be identified in this situation. Therefore, we first tried to find a suitable dye which would stain the arachnoid trabeculae, which would not need acidification of the SAS, and would not fade in acetone during the dehydration procedure. We found that haematoxylin Gill's No.2 or No.3 stain was very stable in acetone. However, Gill's No.3 is such a strong staining solution that all the structures in the SAS were stained as dark blue in a very short incubation period (less than 1 minute). Thus the structures in the SAS became indistinguishable. Haematoxylin Gill's No.2 is optimal for the arachnoid trabeculae staining because its incubation time is easily controlled and it is stable during E12 sheet plastination procedures.
Before plastination, it is also important to fill the SAS with a transparent material to hold the structures in a natural state and prevent any damage to these structures during the sheet plastination procedures. Epoxy resin E20 and 5,10 and 20% gelatine solutions with or without Arabic gum have been tried in this study. Translucent epoxy resin E20 was supposed to be an ideal SAS filling material because red-coloured epoxy resin E20 was used to perfuse arteries in our initial trial and demonstrated a very good vascular filling (figure 2a). However the viscosity of pure E20 is very high and needs to be diluted in acetone. The acetone diluted epoxy resin became milky white in the SAS when cured. This was caused by the residual water in the SAS and the water extracted by the acetone in the diluted epoxy resin. Moreover, it seems that the change of translucent epoxy resin is irreversible. The cured milky white resin in the SAS of the specimen still showed milky white after dehydration in acetone. A mixture of gelatin and Arabic gum recommended by Brasil and Schneider to fill the SAS lost its transparency after dehydration. Twenty percent gelatin solution could provide only a semitransparency after dehydration. Five percent gelatin solution offers an ideal transparency but it will not become solid at room temperature and, most importantly,
cannot provide sufficient support for the contents of the SAS. Based on these findings, we found that 10% gelatin solution is the best selection for SAS filling. It can preserve the SAS in a relatively natural state, provide enough protection for the contents of the SAS during the plastination procedures and still show a satisfactory transparency after dehydration. Under a stereomicroscope, a few fine web-like structures can be seen in some regions of the SAS (figure 2b) and are presumably derived from the gelatin. However, these fine artefactual structures can be easily distinguished from the arachnoid trabeculae as they appear yellowish while the stained arachnoid trabeculae show purple-blue.
The purpose of perfusing vessels with coloured filling materials was to distinguish the small transcisternal arteries and veins from the fine arachnoid trabeculae. As a result of our modification, the small arteries appear red, the small veins blue, and the arachnoid trabeculae purple-blue. These coloured structures are readily distinguishable under the stereomicroscope. It was found that colored epoxy resin E20 is suitable for vascular filling. However, gelatin was found to be unsuitable: upon injection of gelatin into the vascular lumen, many small gaps occurred due to gelatin shrinkage and breakage after dehydration. This may explain why the small perforating vessels are not observable in brain tissue that has been perfused with colored gelatin.
A minor technical point is perhaps worthy of note. Rather than washing out the venous blood prior to fixation, it may be preferable to leave the blood in the veins, so that after plastination, the small transcisternal veins appear almost black and thus easily differentiated from fine arachnoid trabeculae and transcisternal arteries.
We believe that the technique reported in this paper, which combines E12 sheet plastination with the special treatments described will greatly facilitate cisternal anatomy studies and provide a new approach for examining structures in the subarachnoid space at both macroscopic and microscopic levels.
The authors wish to thank Mr. Russell Barnett for his help in E12 sheet plastination technique, Mr. Brynley Crosado for his help in cadaver fixation.
Brasil AVB, Schneider FL: Anatomy of Liliequist's membrane. Neurosurgery 32(6): 956-961, 1993. Cook P: Sheet plastination as a clinically based teaching aid at the University of Auckland. Acta Anat 158: 33-36, 1997.
https://doi.org/10.1097/00006123-199306000-00012
Grondin G, Olry R: Current Plastination Index 1996. Publication of the International Society for Plastination. Universite du Quebec a Trois-Rivieres, Quebec, Canada, 1996.
Matsuno H, Rhoton Jr AL, Peace D: Microsurgical anatomy of the posterior fossa cisterns. Neurosurgery 23(1): 58-80, 1988.
https://doi.org/10.1097/00006123-198807000-00012
Vinas FC, Fandino R, Dujovny M, Chaverz V: Microsurgical anatomy of the supratentorial arachnoidal trabecular membranes and cisterns. Neurological Research 16: 417-424, 1994.
https://doi.org/10.1080/01616412.1994.11740266
Vinas FC, Dujovny M, Frandio R, Chavez V: Microsurgical anatomy of the infratentorial trabecular membranes and subarachnoid cisterns. Neurological Research 18: 117-125, 1996a.
https://doi.org/10.1080/01616412.1996.11740389
Vinas FC, Dujovny M, Frandio R, Chavez V: Microsurgical anatomy of the arachnoidal trabecular membranes and cisterns at the level of the tentorium. Neurological Research 18: 305-312, 1996b.
https://doi.org/10.1080/01616412.1996.11740426
von Hagens G, Tiedemann K, Kriz W: The current potential of plastination. Anat Embryol 175: 411-421, 1987.
https://doi.org/10.1007/BF00309677
Yasargil MG, Kasdaglis K, Jain KK, Weber HP: Anatomical observation of the subarachnoid cisterns of the brain during surgery. J Neurosurg 44: 298-302, 1976.
https://doi.org/10.3171/jns.1976.44.3.0298
Plastination Workshop and 5th Interim Meeting of the International Society for Plastination. The University of Tennessee, Knoxville, TN. Page 22, 1997.